

Modern sequencing technology produces data at a rate which has long exceeded the ability of humans to manually investigate without the aid of computational and algorithmic tools. In addition to the sheer volume of information, viral metagenomes (viromes) are dominated (∼60-90%) by sequences without significant similarity to known organisms in genomic databases, reducing the power of these analyses to investigate viral ecology and diversity. New methods are constantly being developed to combat this ‘unknowns’ problem. Here you will find software created and maintained by the Sullivan Lab and its former members.
 So you have a viral metagenomic sequence, what next?
So you have a viral metagenomic sequence, what next?
Find out more on our Informatics page. To meet the growing needs of the broader community of viral researchers, tools and important datasets need to coexist in a common cyberinfrastructure. iVirus and iMicrobe were developed to support this need leveraging the CyVerse Cyberinfrastructure (iVirus paper here). These projects are led by and/or in collaboration with Bonnie Hurwitz, Assistant Professor, University of Arizona.
Trouble with iVirus? To date, apps often break with Cyverse updates. However, as of Jan 2017, apps should be more robust due to Ben Bolduc’s heroic efforts to convert everything to Singularity. Additionally, CyVerse does not handle documentation well, so all our iVirus documentation is available through protocols.io. If you know Ben, please thank him for his hard (and often thankless) work keeping things running behind the scenes !
Navigate to:
– Bioinformatics tools
– Scripts
– Lab members involved in software development
Tools Recommendations from the Center of Microbiome Science
Community Available Research Tool Resources
To meet the growing needs of the broader community of viral researchers, tools and important datasets need to coexist in a common cyberinfrastructure. iVirus is currently being developed to support this need. Leveraging the preexisting cyberinfrastructure of the CyVerse Collaborative, iVirus lays the foundation for ongoing development of shared resources for viral datasets, metadata, and tools by the viral community. It will allow for comparative metagenomic analyses across diverse environments to identify new genes and function. Moreover, new tools will be captured in the cyberinfrastructure where they can be immediately utilized, and continually developed and adapted by the community to keep pace with rapid tool innovation. iVirus overview
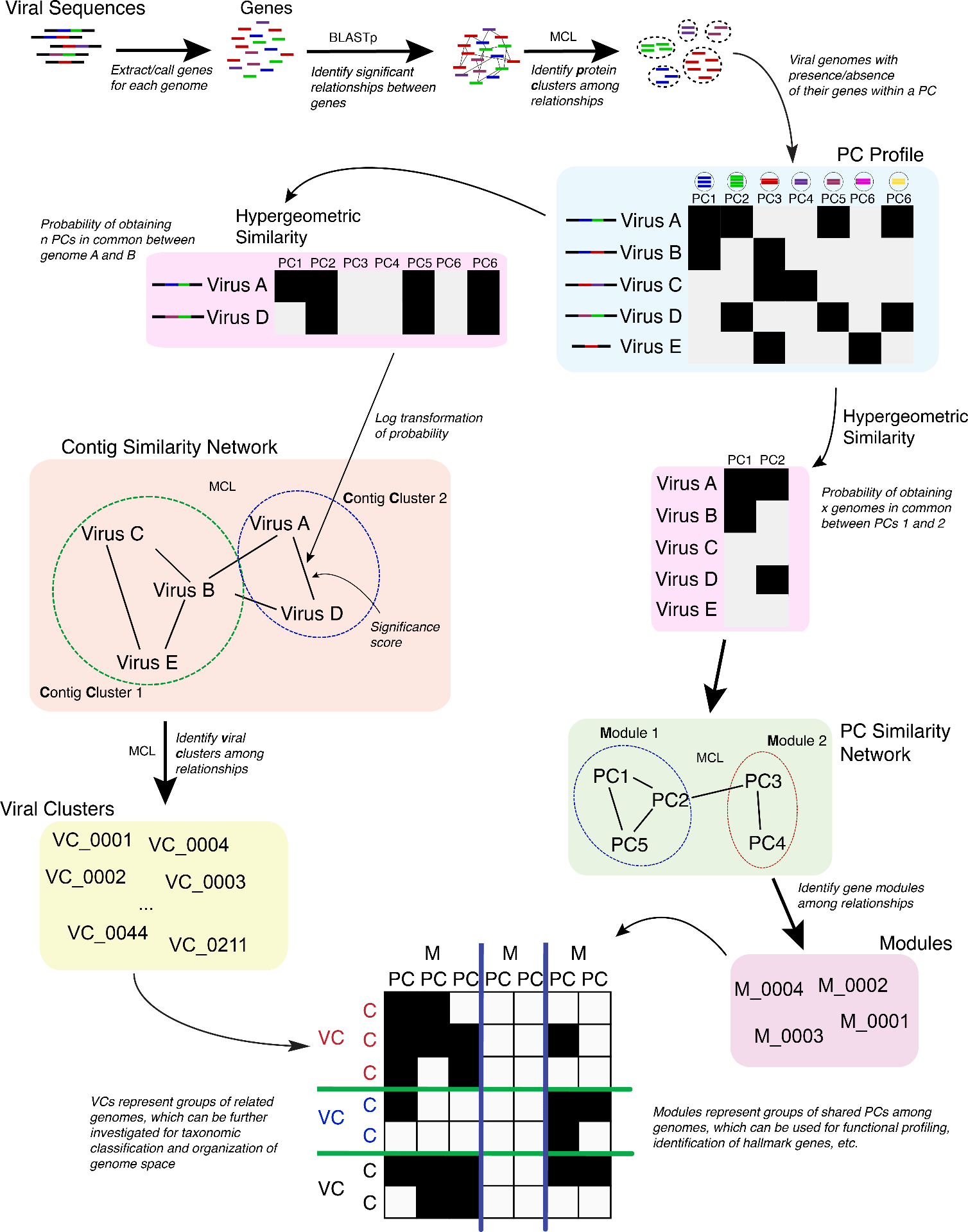
Scripts
Sullivan Lab Code Repository
A Bitbucket repository containing scripts used in by the Sullivan Lab.
Bioinformatics Wiki
Public portion of a lab wiki containing scripts released from the Sullivan Lab.
Hurwitz Scripts
Github repository containing scripts used in Bonnie Hurwitz’s K-mer analysis.
Roux Scripts
Github repository containing scripts used in Simon Roux analysis.