Energy allocation and the function of sleep
Metabolic rate reduction has been considered the mechanism by which sleep conserves energy, similar to torpor or hibernation. This mechanism of energy savings is in conflict with the known upregulation (compared to wake) of diverse functions during sleep and neglects a potential role in energy conservation for repartitioning of biological operations by behavioral state. We refer to this metabolic repartitioning as state-dependent division of labor (DOL). We have developed a mathematical model based on relative rates of energy deployment for biological processes upregulated during either wake or sleep. We quantified the relative contributions toward energy conservation from DOL, metabolic rate reduction, sleep quota and the circadian system, in relation to a continuous wake condition. Our calculations show that coupling of metabolic functions with behavioral state provides comparatively greater energy savings than the measured decrease in metabolic rate, suggesting that actual energy savings derived from sleep are much greater than previous estimates. A combination of state-dependent DOL and modest metabolic rate reduction during sleep can enhance energy savings beyond what is achievable through DOL alone. We propose that state-dependent resource allocation underpins both sleep homeostasis and the optimization of daily energy conservation across species. This new paradigm identifies an evolutionary selective advantage for the upregulation of central and peripheral biological processes during sleep, presenting a unifying construct to understand sleep function.
Brain Physiology – Serotonin and Levodopa
Parkinson’s disease has been traditionally thought of as a dopaminergic disease in whichcells of the substantia nigra pars compacta (SNc) die. However, accumulating evidence implies an important role for the serotonergic system in Parkinson’s disease in general and in physiological responses to levodopa therapy, the first line of treatment. We used a mathematical model to investigate the consequences of levodopa therapy on the serotonergic system and on the pulsatile release of dopamine (DA) from dopaminergic and serotonergic terminals in the striatum.
There are two key ideas that are necessary for understanding the complicated and important connections between the serotonin (5HT) system and the dopamine (DA) system. The first idea is the recognition of the similarities in the synthesis pathways of 5HT and DA in 5HT and DA terminals. The second is volume transmission, which is discussed in a different tab and further below. The following figures show schematically the synthesis, vesicular packaging, release into the extracellular space, reuptake, and control by autoreceptors of DA and 5HT.

DA is synthesized from the amino acid tyrosine (try) that crosses the blood-brain barrier and is taken up by DA nerve terminals by the L-transporter. In the DA terminal, the enzyme tyrosine hydroxylase (TH) adds an OH group to tyr making levodopa (l-dopa). We will abbreviate levodopa by l-dopa and by LD. The amino acid decarboxylase (AADC) cuts off the carboxyl group to make DA, which is indicated by cda, cytosolic DA. The monoamine transporter (MAT) packages cda into vesicles making vda. When the action potential arrives a sequence of events, including Ca++ influx, causes some vesicles to move to the boundary of the terminal and release their contents into the extracellular space.
The extracellular DA, eda, is taken back up into the cytosol by the dopamine transporter (DAT). Extracellular DA also binds to DA autoreceptors (auto) that inhibit synthesis and release. This is clearly a control mechanism intended to stabilize the concentration of eda. Of course, the actual situation is more complicated, for example, cda itself inhibits TH and eda can be taken up by glial cells.
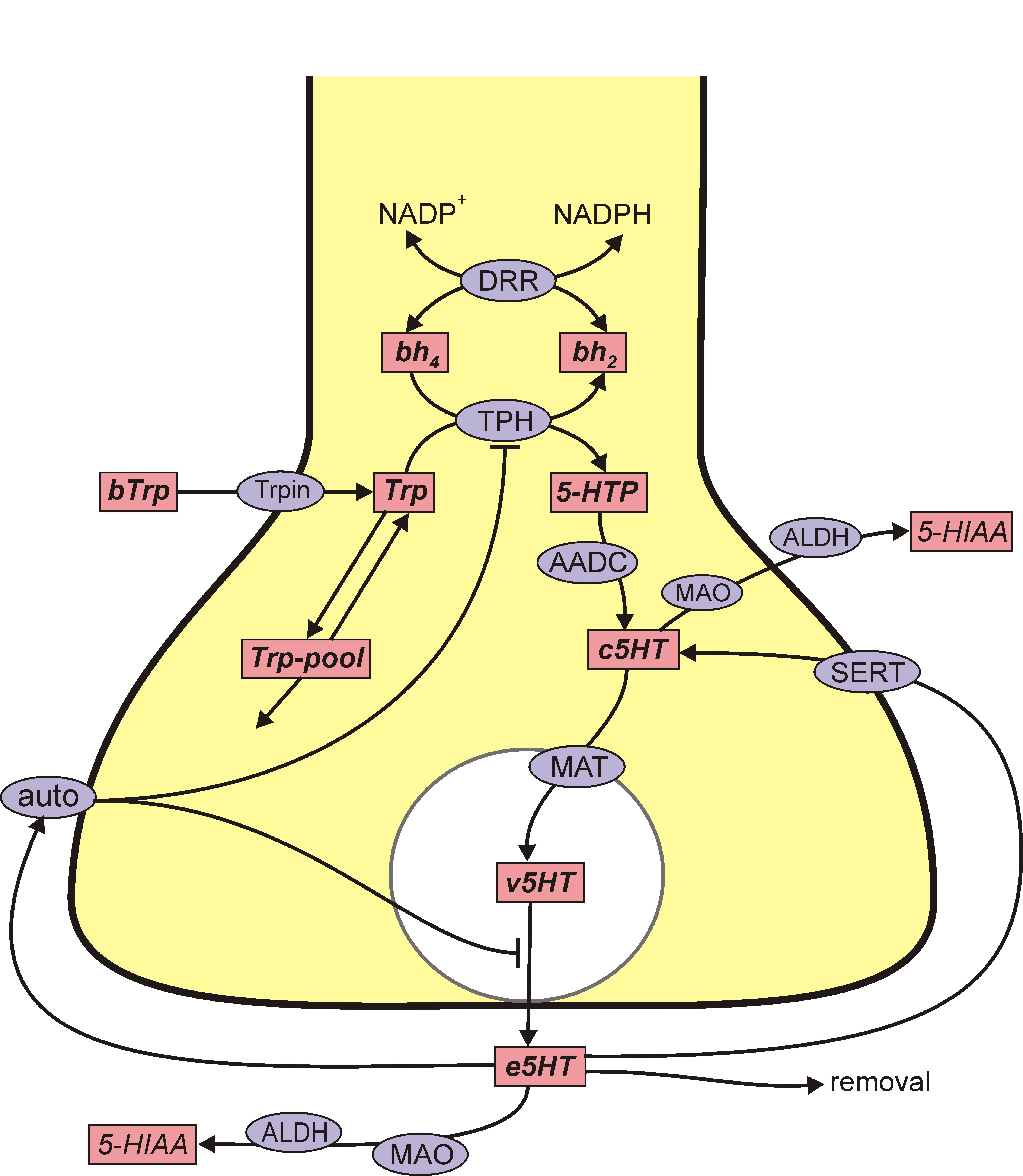 The situation for 5HT is remarkably similar. 5HT is synthesized from the amino acid tryptophan (tryp) that crosses the blood-brain barrier and is taken up by 5HT nerve terminals by the L-transporter. In the 5HT terminal, the enzyme tryptophan hydroxylase (TPH) adds an OH group to tryp making 5-htp. The enzyme amino acid decarboxylase (AADC) cuts off the carboxyl group to make 5HT, which is indicated by c5ht, cytosolic 5HT. The monoamine transporter (MAT) packages c5ht into vesicles making v5ht. When the action potential arrives a sequence of events, including Ca++ influx, causes some vesicles to move to the boundary of the terminal and release their contents into the extracellular space. The extracellular 5HT, e5ht, is taken back up into the cytosol by the serotonin transporter (SERT). Extracellular 5HT also binds to 5HT autoreceptors (auto) that inhibit synthesis and release.
The situation for 5HT is remarkably similar. 5HT is synthesized from the amino acid tryptophan (tryp) that crosses the blood-brain barrier and is taken up by 5HT nerve terminals by the L-transporter. In the 5HT terminal, the enzyme tryptophan hydroxylase (TPH) adds an OH group to tryp making 5-htp. The enzyme amino acid decarboxylase (AADC) cuts off the carboxyl group to make 5HT, which is indicated by c5ht, cytosolic 5HT. The monoamine transporter (MAT) packages c5ht into vesicles making v5ht. When the action potential arrives a sequence of events, including Ca++ influx, causes some vesicles to move to the boundary of the terminal and release their contents into the extracellular space. The extracellular 5HT, e5ht, is taken back up into the cytosol by the serotonin transporter (SERT). Extracellular 5HT also binds to 5HT autoreceptors (auto) that inhibit synthesis and release.
All cells in the body take up tyrosine and tryptophan. The only difference between DA neurons and 5HT neurons is that DA neurons express the enzyme TH and thus make DA and 5HT neurons express TPH and thus make 5HT. As we will explain, this distinction is eliminated in 5HT neurons when one gives a dose of levodopa.
 In Parkinson’s disease, the DA cells in the substantia nigra pars compacta (SNc) die. These cells project to the striatum where low levels of DA cause dysfunction in the motor system. DA does not cross the blood-brain barrier because it doesn’t have a carboxyl group and is not recognized as an amino acid. However, its precursor, l-dopa, still has the carboxyl group and does cross the blood-brain barrier. Thus, the idea of levodopa therapy is to fill the remaining DA terminals in the striatum with l-dopa so that these remaining terminals will release more DA into the extracellular space when action potentials arrive, compensating for the DA terminal loss caused by cell death in the SNc.
In Parkinson’s disease, the DA cells in the substantia nigra pars compacta (SNc) die. These cells project to the striatum where low levels of DA cause dysfunction in the motor system. DA does not cross the blood-brain barrier because it doesn’t have a carboxyl group and is not recognized as an amino acid. However, its precursor, l-dopa, still has the carboxyl group and does cross the blood-brain barrier. Thus, the idea of levodopa therapy is to fill the remaining DA terminals in the striatum with l-dopa so that these remaining terminals will release more DA into the extracellular space when action potentials arrive, compensating for the DA terminal loss caused by cell death in the SNc.
L-dopa is taken up into all cells by the L-transporter, just like tyr and tryp. When l-dopa is taken up into 5HT terminals the enzyme AADC cuts off the carboxyl group to make cda, which is then packaged into vesicles by MAT. Thus vesicles in the 5HT neurons are filled with both 5HT and DA, and when the action potential arrives both are released into the extracellular space. There is a large dense projection of 5HT neurons in the dorsal raphe nucleus to the striatum. So, during a dose of levodopa, the 5HT neurons release a large pulse of DA into the striatum.
All the aspects of this story have been verified experimentally in the last 10 years. Experiments have verified that 5HT neurons can store and release DA in vivo and in vitro. In levodopa treatment of a hemiparkinsonian rat, striatal extracellular DA decreased substantially when the serotonergic system was lesioned. Glial cells also express AADC and so could contribute to the conversion of LD to DA, but experiments using reserpine to block vesicular packaging showed a great reduction of extracellular DA, suggesting that most of the levodopa-derived DA is released by exocytosis of vesicles rather than by glia, at least at physiological levels of levodopa administration. It has also been shown that 5HT1a autoreceptor agonists (that decrease RN firing) and 5HT1b autoreceptor agonists (that decrease release at 5HT terminals) both lower extracellular DA in the striatum in a dose-dependent manner after an LD dose.
The new understanding of 5HT neurons in levodopa therapy has helped to explain a serious side effects of levodopa therapy. Within 5 years of chronic LD treatment, many patients experience a variety of complications (Mouradian et al., 1988). For instance, the length of the therapeutic time window in which a given LD dose relieves PD symptoms gradually shortens and approaches the plasma half-life of LD (wearing-off). Rapid variations in efficacy may occur (on-off fluctuations). Another, particularly troubling, complication of chronic LD therapy is the appearance of involuntary movements (levodopa-induced dyskinesia, LID). These complications increase patients’ disability substantially, pose a therapeutic dilemma, and limit the use of LD.
There is good evidence that large pulses of DA in the striatum are the proximal cause of LID that are seen in long-term dosing. And there is conclusive evidence that these large pulses result from DA release from 5HT neurons in the striatum. Lesioning the 5HT system or giving selective serotonin autoreceptor (5HT1a and 5HT1b) agonists results in a nearly complete elimination of LID.
In order to investigate these phenomena, we created a mathematical model that corresponds to the above diagram. What we discovered was that the size of these large pulses of DA coming from 5HT neurons depended critically on the fraction, f, of SNc cells left alive, which is why there are more and more dyskinesias and Parkinson’s disease progresses. Here is the intuitive explanation. As long as there are lots of SNc cells alive, there will be lots of DA terminals in the striatum with DAT and DA autoreceptors. The DATs take up a lot of the excess DA that comes from the 5HT neurons and the DA autoreceptors restrict DA release from the DA terminals when the extracellular DA concentration is high. However, as the fraction of SNc cells left alive gets smaller and smaller these two control mechanisms have less and less effect. The DA from 5HT neurons is released much faster causing high pulses of DA in the striatum and dyskinesias, and in addition the extra DA created by the levodopa dose is used up much faster shortening the period of efficacy of the LD dose.
 It has been shown in animals that during an LD dose the 5HT released in projection regions is about half of normal. It is interesting to speculate whether this might be a partial cause of depression in some Parkinson’s patients.
It has been shown in animals that during an LD dose the 5HT released in projection regions is about half of normal. It is interesting to speculate whether this might be a partial cause of depression in some Parkinson’s patients.
Legend. (A) Shows the time course of extracellular DA in the striatum for different values of f, the fraction of SNc cells left alive. As f declines the curves get higher because there are fewer DATs to take up the DA released by 5HT neurons. However, when f is very small the peaks decline because removal mechanisms such as catabolism, diffusion, and uptake into glial cells become more important. The dashed black horizontal line in (A) represents the level of extracellular DA needed in the striatum for anti-Parkinsonian effects. (B) Reproduces the first three curves from (A) (solid curves, f = 1, f = 0.2, f = 0.1). The dashed curves in the same color show the amount of extracellular DA in the striatum that comes from the DA neurons. For a normal individual (f = 1) the DA neurons contribute approximately 60%, but as SNc cells die (f = 0.2, f = 0.1) most of the DA comes from the 5HT neurons. (C) Shows that the amount of time that extracellular DA stays above the therapeutic level (the dashed black line in (A)) declines as PD progresses until it becomes approximately 2 h.
Brain Physiology – Homeostasis of dopamine
Neurotransmitters provide the mechanism by which electrical signals are communicated from one neuron to the next. However, there is strong evidence that in many cases it is the concentration of a neurotransmitter in the extracellular space of a nucleus that affects electrophysiological neurotransmission by other neurotransmitters (volume transmission). This raises several natural questions. What are the mechanisms by which the extracellular concentrations of neurotransmitters are controlled? How do neurotransmitters in the extracellular space affect synaptic transmission by other neurotransmitters? How robust are these mechanisms in the face of polymorphisms in the enzymes affecting synthesis, release, and reuptake of neurotransmitters? How are dysfunctions in these control mechanisms related to neurological and neuropsychiatric diseases? In this section we briefly describe our work on several of these questions.
Passive stabilization of DA in the striatum. Parkinson’s disease is a neurodegenerative disorder associated with cell loss from the substantia nigra pars compacta (SNc). The dopaminergic cells of the SNc send projections to the striatum where the loss of dopaminergic tone is thought to be the main cause of tremor and other motor symptoms of Parkinsonism. An interesting and important feature of the disease is that symptoms do not appear until a very large percentage (75%-90%) of SNc cells have died and therefore this feature has been the focus of much experimental and clinical investigation. Experiments with animal models [6e8] have shown that although striatal tissue content of dopamine declines more or less proportionally to cell death in the SNc, the extracellular concentration of dopamine (EDA) in the striatum remains near normal until more than 85% of SNc neurons have died. This is widely believed to be the reason that symptoms do not appear until very late in the degeneration of the SNc.
What is the basis of this remarkable homeostasis of striatal EDA in the face of progressive cell death in the SNc?
Some researchers proposed that the nigrostriatal system adapts to cell death to maintain extracellular DA level by increasing DA synthesis in the living terminals or by sprouting more terminals. But in 2003 Bergstrom and Garris (J. Neurochem 87,1224-36) proposed a very simple explanation that they called “passive stabilization’’ and provided experimental evidence for it. The extracellular concentration of DA depends on the balance between release of DA and reuptake of DA by the dopamine transporters (DATS). If half of the SNc cells die, there will be only half as much release, but there will also be only half as much reuptake, so the concentration of DA in the extracellular space should remain the same.
We used our mathematical model of a DA terminal to investigate the proposal of Bergstrom and Garris. Notice that their hypothesis does not explain why passive stabilization breaks down when, f, the fraction of SNc cells left alive gets small. We believe that passive stabilization breaks down at small f because there is always some removal of DA from the system in the extracellular space by uptake into glial cells and blood vessels or simply diffusion out of the tissue. As the number of DA terminals in the striatum gets smaller, these removal effects get proportionally larger because the reuptake DATs become sparser and sparser. This hypothesis was confirmed and explained by our mathematical modeling. The figure above shows the concentration of DA in the striatum in the model as a function of f, the fraction of SNc cells left alive. One can see that the passive stabilization effect of Bergstrom and Garris keeps the extracellular DA concentration quite constant until approximately 80% of the SNc cells have died. As even more cells die the concentration drops to zero because the removal effects dominate more and more. The dashed curves show that the passive stabilization depends on the dopamine transporters.
Control of extracellular DA by autoreceptors. Under the tab “constructing models’’ we gave as an example our 2009 model of a DA terminal including synthesis, packaging into vesicles, release, and reuptake via the DATs. We also included the effects of the DA autoreceptors that sense the DA concentration, EDA, in the extracellular space. When DA gets higher than normal, the autoreceptors inhibit synthesis and release of DA, and when DA gets lower than normal this inhibition is withdrawn stimulating synthesis and release. Thus the autoreceptors act to modulate EDA against both long term and short term perturbations such as changes in the supply of tyrosine or changes in firing rate. The mechanisms by which EDA affects synthesis and release via the autoreceptors are mostly unknown and an important topic of current research that involves difficult questions in cell biology. The control of DA in the extracellular space is also affected by other neurotransmitters. For example, there is dense serotonergic projection to the striatum from the dorsal raphe nucleus (DRN). The released 5HT binds to 5HT receptors on DA terminals and increases DA release when the SNc cells fire.
Homeostasis of DA and cryptic genetic variation. An important field of study in the past 15 years has been to quantify the effects of gene polymorphisms on the proteins that are important for the dopaminergic system, for example, tyrosine hydroxylase (TH) or the dopamine transporter (DAT). Typically, these experiments are done in test tubes or in vitro and the polymorphisms often have large quantitative effects on the function of the proteins. And, it is very tempting to conclude that the polymorphisms are therefore the “causes’’ of various neurological or neuropsychiatric diseases. However, in vivo there are many control mechanisms  (two are discussed above) that buffer the DA concentration in the extracellular space against perturbations in the DA system. We pointed this out already in our 2009 paper, but the point is made dramatically by the two dimensional surface taken from our 2014 paper. The surface shows the extracellular DA concentration (z-axis) at steady state as a percentage of normal as a function of the activity of tyrosine hydroxylase and the efficacy of the dopamine transporter computed from our model. In both cases, 1 indicates normal activity for TH and DAT. The large white dot on the surface is wild type, the concentration of EDA when TH and DAT have their normal activities. The smaller white dots on the surface indicate points that correspond to common polymorphisms (homozygotes and heterozygotes) in the human population taken from the table. Notice that all the white dots lie on the flat part of the surface where the polymorphisms cause only very modest changes in extracellular DA despite the fact that they cause large changes in protein activity. This is the effect of the autoreceptors. It is quite amazing that all these polymorphisms all lie on the flat part of the surface. Presumably, if they didn’t, they would have been selected against and would not be common in the human population. This example shows why one has to be very careful about jumping to physiological conclusions from test tube or in vitro experiments.
(two are discussed above) that buffer the DA concentration in the extracellular space against perturbations in the DA system. We pointed this out already in our 2009 paper, but the point is made dramatically by the two dimensional surface taken from our 2014 paper. The surface shows the extracellular DA concentration (z-axis) at steady state as a percentage of normal as a function of the activity of tyrosine hydroxylase and the efficacy of the dopamine transporter computed from our model. In both cases, 1 indicates normal activity for TH and DAT. The large white dot on the surface is wild type, the concentration of EDA when TH and DAT have their normal activities. The smaller white dots on the surface indicate points that correspond to common polymorphisms (homozygotes and heterozygotes) in the human population taken from the table. Notice that all the white dots lie on the flat part of the surface where the polymorphisms cause only very modest changes in extracellular DA despite the fact that they cause large changes in protein activity. This is the effect of the autoreceptors. It is quite amazing that all these polymorphisms all lie on the flat part of the surface. Presumably, if they didn’t, they would have been selected against and would not be common in the human population. This example shows why one has to be very careful about jumping to physiological conclusions from test tube or in vitro experiments.
This surface is a perfect example of cryptic genetic variation in which large variation in genes (gene products), that is, TH and DAT, produce very little variation in a phenotypic variable, the extracellular DA concentration. It should be kept in mind that the actual situation is much more complicated than this two dimensional surface in three space would lead one to believe. There are many other variables, both genetic variables (for example a polymorphism in the monoamine transporter) or phenotypic variables (for example the 5HT concentration, see below) to could affect the shape of this surface. The “real’’ surface is a high dimensional surface in a high dimensional space. Nevertheless this surface does tell us a lot, and it is interesting to think about the people who have the 15% mutation in TH. They are the ones to the right sitting at the edge of the cliff where DA drops to zero. Interestingly, these genotypes sometimes show a dystonia, involuntary muscle contractions that affect posture, brought about by low levels of extracellular DA that can be alleviated by levodopa. So, one could say that their position at the edge of the cliff (that is, having the 15% TH polymorphism) predisposes them to the dystonia. Some of them are pushed over the cliff by other variables not pictured and thus show the dystonia. The job of a precision medicine provider would be to advise a patient with the 15% TH polymorphism how to flatten the region around where they lie on the surface and thus to avoid being pushed over the cliff by other variables. In our simulations, the region around these individuals gets flatter if one increases the strength of the autoreceptor effect.
Does 5HT stabilize DA in the striatum? The dopaminergic cells in the SNc project to the striatum where they stimulate spiny neurons in direct pathway from the Cortex to the thalamus and they inhibit spiny neurons in the indirect pathway. The direct pathway stimulates the thalamus and the indirect pathway inhibits the thalamus. The simplest conceptual model of Parkinson’s disease is that when SNc cells die the extracellular DA concentration drops in the striatum removing excitation from the direct pathway and removing inhibition from indirect pathway, thus tipping the balance towards the indirect pathway that inhibits the thalamus. The resulting inhibition of the thalamus cause the inability to initiate movement. But, what does this have to do with 5HT? There is a dense 5HT projection to the striatum from the dorsal raphe nucleus (DRN) and it is known that increased concentrations of 5-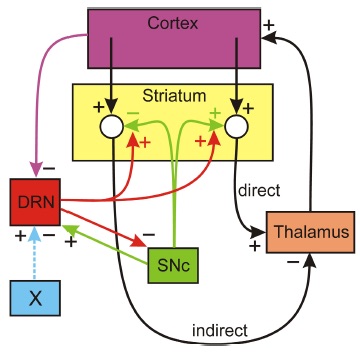 HT in the striatum facilitate the release of DA from the dopaminergic projections from the SNc. Thus 5HT could partially compensate for cell loss in the SNc if the DRN fires more when SNc cells die. There is a plausible mechanism by which this could happen. When SNc cells die there is more inhibition of the thalamus (as discussed above). Since projections from the thalamus excite cortical neurons there will be less stimulation of the cortex. There are numerous projections from higher brain regions to the DRN; in particular, there are inhibitory projections from medial prefrontal cortex and if these projections fire less, then the DRN neurons would fire more. This is speculative, but gives a plausible mechanism by which the 5HT projection from the DRN to the striatum acts to stabilize DA in the striatum. That this idea would work is supported by a simple mathematical model, but not enough is known about the projections from the thalamus to the cortex and from the cortex to the DRN to be sure of the anatomy.
HT in the striatum facilitate the release of DA from the dopaminergic projections from the SNc. Thus 5HT could partially compensate for cell loss in the SNc if the DRN fires more when SNc cells die. There is a plausible mechanism by which this could happen. When SNc cells die there is more inhibition of the thalamus (as discussed above). Since projections from the thalamus excite cortical neurons there will be less stimulation of the cortex. There are numerous projections from higher brain regions to the DRN; in particular, there are inhibitory projections from medial prefrontal cortex and if these projections fire less, then the DRN neurons would fire more. This is speculative, but gives a plausible mechanism by which the 5HT projection from the DRN to the striatum acts to stabilize DA in the striatum. That this idea would work is supported by a simple mathematical model, but not enough is known about the projections from the thalamus to the cortex and from the cortex to the DRN to be sure of the anatomy.