Multiple sclerosis (MS) is an immune-mediated, demyelinating disease, responsible for deleterious consequences within the central nervous system (CNS). Progressive inflammation, demyelination of myelin sheaths, plaque formation, and axonal degeneration are the processes that characterize MS. Variations involving the cellular mechanics driving these processes exist. However, MS usually begins with an immune-mediated inflammatory response (Lassman, 2013).
T lymphocytes, specifically CD4+ (T helper cells) and CD8+ (cytotoxic T cells), recognize myelin as foreign and attack it, initiating a damaging, inflammatory response (Lassman, 2013; McCance & Huether, 2014; Tullman, 2013). T cells are activated by antigen presenting cells (APCs), which when activated, cross into the CNS through a disrupted blood brain barrier (BBB). Passage is facilitated by integrins, which are expressed by activated T cells, allowing the adhesion and passage of immune cells through the BBB. CD4+ differentiates into T-helper type (Th) cells, which secrete specific types of cytokines. Th1 is pro-inflammatory and Th2 is anti-inflammatory. Th17 promotes inflammation with secretion of interleukin-17. CD8+ cells kill neuroglial cells, expose axons, and activate oligodendrocyte death. B cells and microglia become activated and the influx of macrophages, eosinophils, and neutrophils occur. Antigen-activated B cells differentiate into antibody-secreting plasma cells, serve as APCs to T-cells, and secrete inflammatory cytokines. Infiltrating B cells and myelin antibodies activate complement, which promotes inflammation during acute phases and is neuroprotective during periods of relapse (Lassman, 2013; McCance & Huether, 2014; Tullman, 2013).

(Nature reviews, 2009)
The immune-mediated inflammatory response ultimately leads to the destruction of neuroglial cells and demyelination. The body attempts to repair damages through remyelination. The repetitive process of demyelination and remyelination leads to scarring and plaque formation, characteristic of this disease process (Cross, A., Cross, K., & Piccio, L. 2012; Lassman, 2013). Myelin destruction and axonal damage ensues when efficient remyelination overwhelms demyelinating processes. Inflammation and demyelination is exacerbated by the release of neurotoxins and reactive oxygen species. Damage to axonal ion homeostasis, leads to calcium influx, which is proinflammatory and neurotoxic. Activated immune cells produce another neurotoxin, glutamate. The rapid activation of microglia and macrophages leads to an oxidative burst, a major component of mitochondrial dysfunction and demyelination, involving the release of nitric oxide and oxygen free radicals (Cross, A., Cross, K., & Piccio, L. 2012; Lassman, 2013).
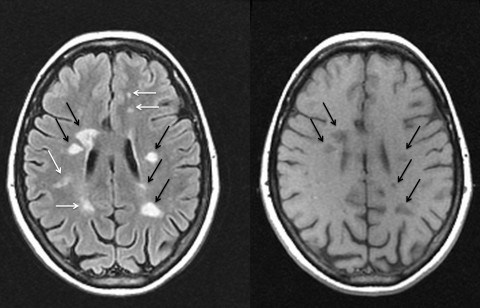
White arrows show areas of active lesions and black arrows show up as persistent “black holes” indicating a more severe degree of tissue destruction. Obtained from http://www.msdiscovery.org/news/news_synthesis/322-more-meets-eye
The presence of scarring and plaque formation seen with an MRI is the primary diagnostic feature of MS (Lassman, 2013). This occurs in all stages of the disease and develops in both the white and grey matter. As disease progresses, diffuse tissue injury will lead to widespread loss of tissue volume, brain atrophy, and dilation of the ventricles (Lassman, 2013).
Clinical manifestations correlate with the portion of the CNS involved and extent of destruction. Most patients begin with a clinically isolated syndrome and a relapsing-remitting MS (RRMS) disease course follows (Cross, A., Cross, K., & Piccio, L. 2012; McCance & Huether, 2014). RRMS often progresses to secondary progressive MS (SPMS) where deficits continue to progress without distinct periods of remission. A small percentage of patients begin with primary progressive MS (PPMS) and demonstrate a steady, progressive course of disease following the initial onset without remission or relapse (Cross, A., Cross, K., & Piccio, L. 2012; McCance & Huether, 2014).
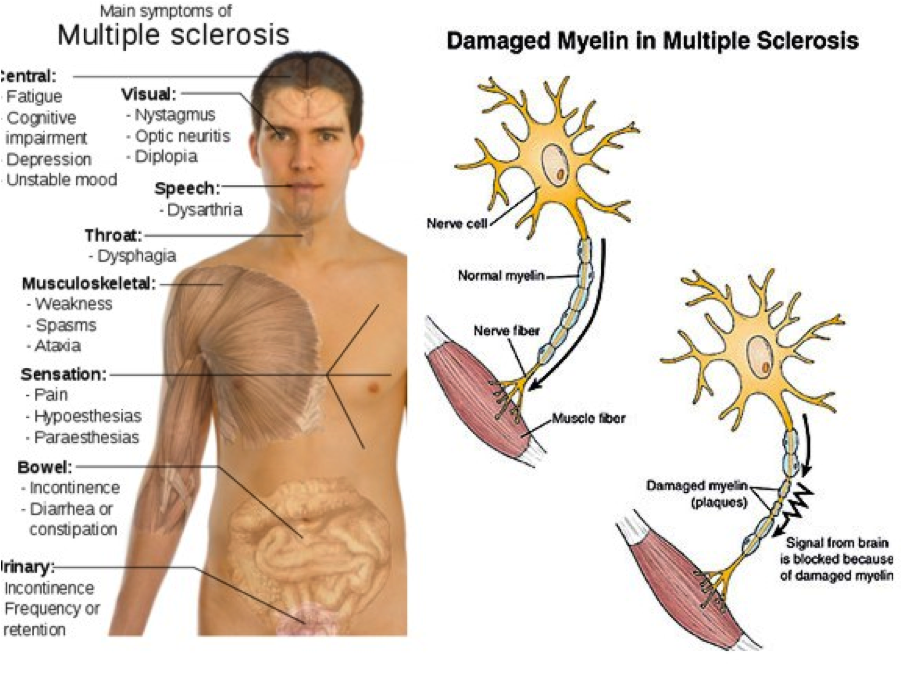
(Multiple sclerosis: causes, 2012)
Optic nerve injury (optic neuritis) can cause a disturbance in central vision (blurring, fogginess, haziness) and/or impaired color perception (Gelfand, 2014; McCance & Huether, 2014). Clinical manifestations seen with brainstem lesions include double vision, internuclear ophthalmoplegia, facial weakness, facial sensory deficits, vomiting, tinnitus, vertigo, dysphagia, dysarthria, and tongue weakness. Cerebellar disturbance can cause motor ataxia, hypotonia, and asthenia. Lesions of the spinal tracts and dorsal column can cause weakness, numbness, stiffness, slowness, and weakness. Major spinal involvement will cause bladder and bowel symptoms where urgency, hesitancy, incontinence, neurogenic impotence, and/or a spastic bladder may occur (Gelfand, 2014; McCance & Huether, 2014).
Fatigue often persists even during inactive phases of the disease and is likely the result of chronic CNS inflammation (Gelfand, 2014). Cognitive impairment (slowed information processing, executive dysfunction, or impaired memory) is caused by brain atrophy and white matter involvement. Depression is also commonly seen, however the cause is unknown (Gelfand, 2014).