
Undergraduate Research
Honors Research Thesis

I successfully defended my Honors Senior Research Thesis! Check out my thesis here: Deciphering the mechanisms of dedifferentiated liposarcoma resistance to targeted therapy.
2021 Autumn Undergraduate Research Festival
Project Title: DEVELOPING AND CHARACTERIZING MODELS OF RESISTANCE TO CYCLIN-DEPENDENT-KINASE 4 (CDK4) INHIBITION IN THE CDK4 AMPLIFIED CANCER, DEDIFFERENTIATED LIPOSARCOMA
Project Mentor: James Chen – Internal Medicine
ABSTRACT
Introduction/Background:
Dedifferentiated liposarcomas (DDLPS) are malignant adipocytic cancers characterized by an amplification of the cell cycle regulatory gene, CDK4. Palbociclib, a CDK4/6-inhibitor, is rapidly becoming a standard of care treatment; however, innate and acquired resistance to the drug limits efficacy in patients with DDLPS. Molecular mechanisms and biomarkers of palbociclib resistance have been posited in non-mesenchymal tumors, but are poorly understood in DDLPS. To this end, we developed palbociclib-resistant DDLPS cell lines derived from several patient DDLPS samples to better characterize these phenotypic and genomic differences.
Methods:
Three human CDK4 amplified DDLPS cell lines (224A, 246, 863) and one liposarcoma control cell line without CDK4 amplification and RB loss (LiSa2) were brought into culture. Developing resistant cells were intermittently treated at palbociclib’s IC25 and surviving cells were allowed to recover before retreatment. Parental lines were grown as controls in tandem with the developing resistant lines to compare molecular changes in response to palbociclib. Cell viability was measured by an XTT assay and protein expression was measured using whole cell lysates for Western blotting.
Results:
Parental IC50 of the three CDK4 amplified DDLPS cell lines ranged from 8-12 µM and the cell line without CDK4 amplification had an IC50 of 12 µM. Resistant cell strains demonstrated minimal changes in the measured IC50. Initial response to palbociclib decreased expression of cell cycle proteins, CDK4 and pRb, and increased Cyclin E, CDK2, and CDK6. After 6 months of treatment, a statistically significant decrease in Cyclin E, CDK2, and pRb, was observed in the resistant cell lines and no changes were seen in CDK4, Cyclin D, and p16. (all p<0.01).
Conclusion:
Here we present the development of novel, palbociclib resistant, DDLPS cell lines. While the initial response to combat palbociclib treatment increased Cyclin E and CDK2, these G1/S proteins are reduced in resistant lines. Resistant cell lines demonstrated decreased protein expression of Cyclin E, CDK2, and pRb. Clinically, DDLPS patients that exhibit innate dysregulation of RB1 or G1/S phase regulators may be more likely to demonstrate resistance to palbociclib. Next-generation sequencing of DNA and RNA expression in parent and resistant lines are underway.
About my research: Treating enzalutamide-resistant prostate cancer
This summer, 24 CSPC/PS-ON fellows and I learned how to use R-studio to analyze a gene data set to understand breast cancer metastasis. At the end of our 10-week internship, we participated in a hackathon-style project. My partner and I had three hours to come up with a solution to enzalutamide-resistant prostate cancer.
The following infographic covers the basics of our proposal. This project was an opportunity to creatively think of an approach to treat drug resistance, consider the limitations of different methods, and develop scientific communication skills, not to come up with solid answers.


About my research: Imaging of pancreatic tumors in 3D to study venous invasion
Click the image to see it move!
INTRODUCTION
Pancreatic cancer remains one of the deadliest solid malignancies, with a 5-year survival rate of approximately 10% in the USA. Patients often do not show symptoms until cancer reaches an advanced stage where cancer can aggressively metastasize (travel to different parts of the body)–the mechanism of which remains unknown. Venous invasion occurs when cancer cells gain access to the veins and is a promising explanation for the development of liver metastases and thus the rapid progression of disease in most patients.
BACKGROUND
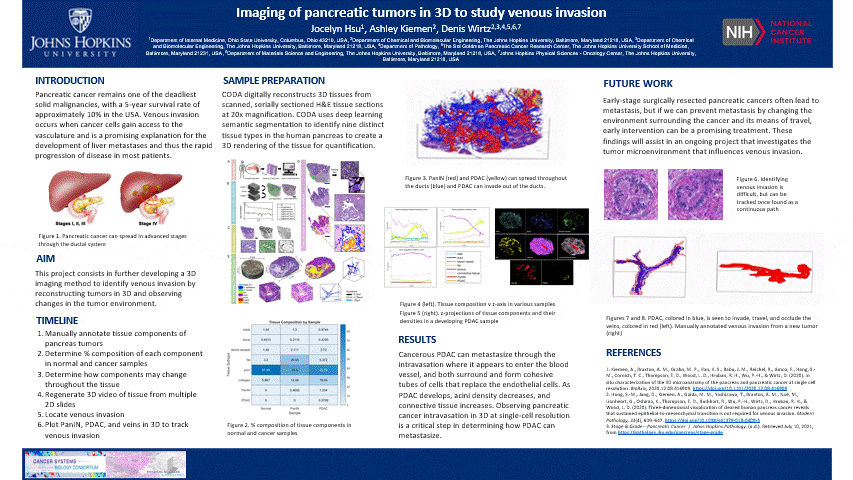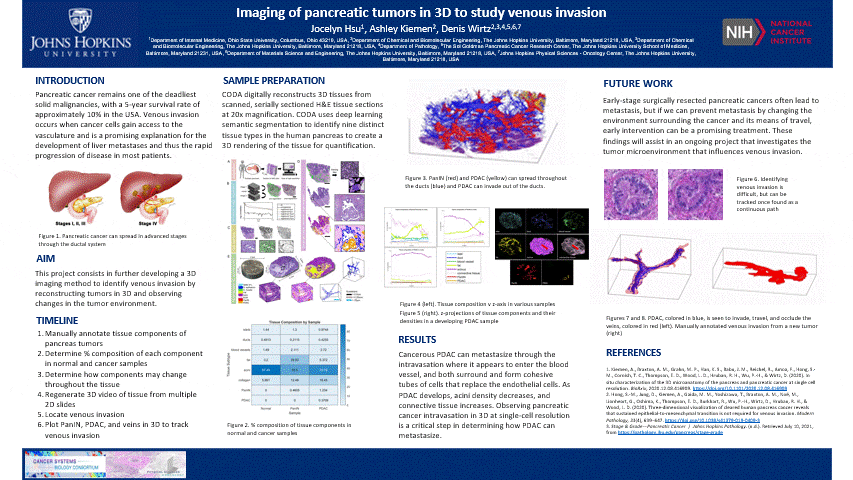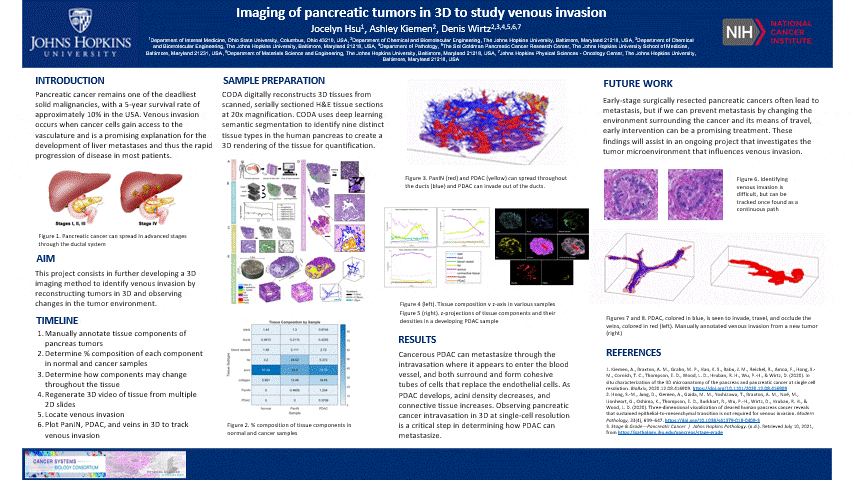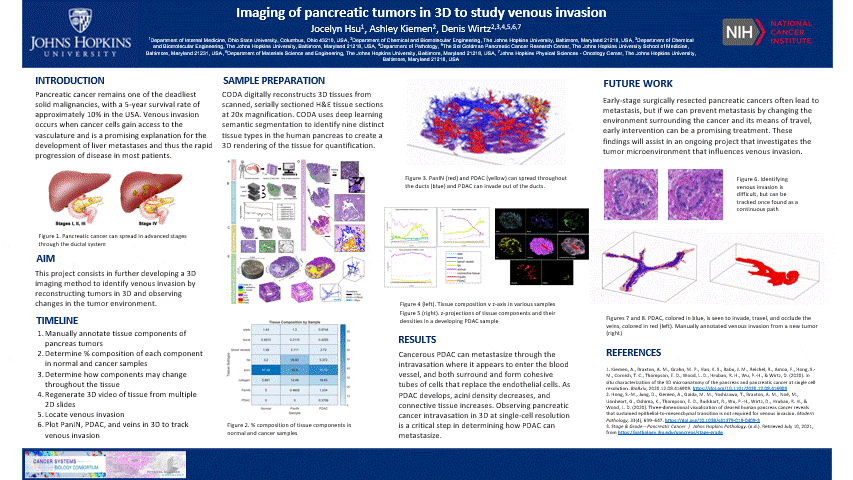
The pancreas is located behind the stomach and underneath the liver. It aids in digestion and blood sugar regulation. Pancreatic cancer develops in stages including the formation of precancerous pancreatic intraepithelial neoplasia (PanIN) which forms as a result of genetic mutations impacting cell-cycle progression, cell division, and cell growth in ductal cells. These mutated cells can multiply and travel through the ductal system until it potentially develops into cancerous pancreatic ductal adenocarcinoma (PDAC).
AIM
This project consists of further developing a new imaging method of tumors in 3D at single-cell resolution named CODA–to be validated and tested by studying venous invasion in pancreatic cancer. CODA is based on novel registration and deep learning algorithms.
Why is 3D imaging important to study venous invasion? As of now, we’ve only been able to look at veins in 2D cross-sections.

- A: If my vein is horizontal, and I cut the biopsy horizontally, then I will see the whole vein. Great!
- B: What if the vein is vertical? If I cut the biopsy horizontally, then I will see a dot–just a section of the vein
- C: Okay, let’s say I can see a dot. Well that means 1 dot == 1 vein. So 2 dots == 2 veins, right? Actually, veins can branch off. So 2 dots could mean we have 1 vein with a branching point
- Conclusion: you cannot determine vein position or branching without looking at the whole thing in 3D. As of now, we’ve only been able to see veins in 2D, so reconstructing full tissues after they’ve been sliced up is really neat
METHODS
- We (not me, but someone else) took a biopsy of a patient’s pancreas and sliced it into multiple horizontal sections every 4 μm. Then they stained it so different pancreas cells are stained different shades of white, pink, blue, and red so it is easier to identify them

- Manually annotate tissue components of pancreas tumors
- I spent my first three weeks circling different components of pancreatic tissue like fat, blood vessels, and ducts to help the computer identify patterns and manually determine what type of cells the tumor image had. “Hey, Computer, these white circles are fat. So if you see one of those white circles, identify that as fat”

- I spent my first three weeks circling different components of pancreatic tissue like fat, blood vessels, and ducts to help the computer identify patterns and manually determine what type of cells the tumor image had. “Hey, Computer, these white circles are fat. So if you see one of those white circles, identify that as fat”
- Determine % composition of each component in normal and cancer samples
- Once the computer identifies the components in the whole image, I want to know how much of the pancreas sample is made of cancer cells or other components
- In the heatmap below, I’ve analyzed normal pancreas (no cancer), PanIN (precancerous), and PDAC (pancreas sample with cancer). We can see that as the cancer develops, acini decreases and collagen increases
- The pancreas is mostly acini. Those are the cells that produce enzymes for digestion. So the cancer may actually kill those important cells… not good!
- Collagen is like the support structure that mainly surrounds blood vessels (and your muscles/skin/bones/tendons). Collagen tends to increase as cancer develops and we believe that stronger, denser collagen provides a shield to protect cancer and allow it to grow without immune cells attacking it

- Determine how components may change throughout the tissue
- Let’s just confirm that collagen increases where cancer develops. Because these numbers are great, but what if collagen was increasing in a corner of the pancreas and had nothing to do with developing cancer.
- This graph plots the % composition of each slice. We can see that as PDAC increases (orange) (take my word, PDAC develops near the 3-4 z-plane, it’s just very minuscule in comparison to the rest of the graph), collagen (pink) also increases and acini (purple) decreases


- Determine how the density of tissue components change
- From Figure 3.5 (blue heatmap), we can see that acini decreases and collagen increases as PDAC develops. But how do we know that these changes are directly related to the formation of PDAC? If we see a large increase in collagen surrounding PDAC and a decrease in acini around PDAC, we can conclude that PDAC is the reason for acini and collagen changes.
- To do this, we can calculate the density of tissue components as a function of distance from PDAC. Density of Collagen = #pixels of Collagen within a certain distance from PDAC / # total pixels within a certain distance from PDAC
- In the graph below, the x-axis is the distance from PDAC and the y-axis is the density. When we’re close to PDAC (x=0), collagen is high and acini are low which matches our previous observations. As PDAC develops, collagen increases, and acini decreases.


- Regenerate 3D video of tissue from multiple 2D slides
- This is nice and all, but let’s see the cool stuff. The blue are ducts that are normal in the pancreas, the red are blood vessels, and the yellow is the cancer
- Click the image!

- Locate venous invasion
- In another tumor sample, we found venous invasion
- When I bring up venous invasion, most people think the cancer will make its way into the vein and travel to another place. BUT the cancer will actually get comfy, enter the vein, and stick to the interior, replicate, and take up the ENTIRE vein. Therefore, cutting off circulation through that vein. It can still travel through the vein and exit to another place
- The blue is cancer and red is the vein. The cancer completely occludes the vein
- Click the image!

- Calculate blood vessel and PDAC length
- Using MATLAB, I was able to calculate lengths of the blood vessel. Notice how the vein branches off instead of remaining a singular tube. We can calculate the length of each of the extensions. If I can calculate the length of the blood vessel and the length of the cancer (PDAC), then I can see if PDAC extends throughout the entire vein. In MATLAB, this figure is measured in pixels, so I included both pixels and mm lengths.

- Using MATLAB, I was able to calculate lengths of the blood vessel. Notice how the vein branches off instead of remaining a singular tube. We can calculate the length of each of the extensions. If I can calculate the length of the blood vessel and the length of the cancer (PDAC), then I can see if PDAC extends throughout the entire vein. In MATLAB, this figure is measured in pixels, so I included both pixels and mm lengths.
- Calculate PDAC occlusion
- Now we know that PDAC can travel a significant length inside the vein, does PDAC take up the entire space inside? or does it only touch the inside vein walls and leave an open tube through the center? The amount of space PDAC takes up inside of the vein is defined as occlusion.
- To determine occlusion, I can use a single slice of the total volume, calculate the area of the PDAC and then calculate the area of the PDAC and the open space inside the PDAC (this is called lumen). For example, if PDAC looked like a straw inside the vein, its area would only be the outer area of the straw and the lumen would be the open space. That is a low occlusion. BUT if PDAC looked like a wooden dowel inside the vein, its area would be the entire circle, and there would be no lumen. The occlusion would be 1.

- Once I calculate the occlusion for each z-plane of the 3D volume, I calculated the average occlusion to be 0.83


FUTURE WORK
Although it is evident that pancreatic cancer can travel through the vascular system, determining factors that inhibit or promote movement will aid in preventing metastasis. Early-stage surgically resected pancreatic cancers often lead to metastasis, but if we can prevent metastasis by changing the environment surrounding cancer and its means of travel, early intervention can be a promising treatment. These findings can also help to stop developing metastasis and attack cancer at locations such as the bloodstream if that is its main source of travel. Concern also rises over the method of travel for noninvasive PanIN through the vascular system and how that can lead to the implementation of several precancerous spots in various organs prior to PDAC formation.
About my research: CDK4-Inhibitor Resistance in Dedifferentiated Liposarcoma
OVERVIEW
My lab uses bioinformatics, a method that uses computer science to interpret biological data, to understand sarcoma. Sarcomas are soft tissue (like fat, nerve sheaths, connective tissue, smooth muscle, and vessels) and bone cancers that make up about 1% of all cancers. The subtype that I work with is dedifferentiated liposarcoma (DDLPS). Lipo- meaning “fat” and dedifferentiated meaning the cells revert from specialized fat-cell properties to STEM-cell properties so that they can replicate.
DDLPS naturally have an amplified expression of a protein, CDK4, which acts like a traffic light during the cell cycle when it connects with its partner, cyclin D. The cell cycle is split up into different phases so the cell has time to prepare nutrients and duplicate its cellular components (G1), duplicate DNA (S), prepare for mitosis (G2), and then divide (M). The first traffic light is between the G1/S phase and when CDK4 is amplified, the light is green, even if the cellular components are not prepared in cancerous cells. Overexpression of CDK4 then leads to uncontrollable cell division and growth.
Therefore, using a CDK4-inhibitor would be an attractive targeted therapy for patients with DDLPS. Unfortunately, while these drugs have positive outcomes, some patients develop resistance or are innately resistant to the drug. My project aims to develop palbociclib (a CDK4-inhibitor) resistant DDLPS cell lines to determine what proteins are changing expression levels to promote resistance. From here, if I know that Protein ABC increases and Protein 123 decreases in resistant cells, then I probably should not give palbociclib to patients who have high ABC and low 123 because they are more likely to develop resistance. I can also use this information to develop combination therapies. If ABC is increased in resistant cells, then maybe we should treat patients with CDK4 and ABC inhibitors as a double-attack.
HYPOTHESIS
 The cell cycle is complex and relies on many protein interactions to activate/deactivate the traffic lights. There are proteins upstream (proteins that can activate/deactivate CDK4) and downstream (CDK4 activates/deactivates those proteins) that we can look at. In the schematic above, p16 is an upstream protein that deactivates CDK4 (the T-shaped arrow means inhibit) so we would expect p16 to decrease in CDK4-inhibitor-resistant cells (if we want to continue replicating, we need CDK4, so we don’t want p16 to inhibit CDK4). On the other hand, if CDK4 is inhibited, we might rely on another CDK-cyclin pair to activate the traffic light. CDK2 and Cyclin E can do that, so I expect both of those downstream proteins to increase to accommodate for inhibited CDK4.
The cell cycle is complex and relies on many protein interactions to activate/deactivate the traffic lights. There are proteins upstream (proteins that can activate/deactivate CDK4) and downstream (CDK4 activates/deactivates those proteins) that we can look at. In the schematic above, p16 is an upstream protein that deactivates CDK4 (the T-shaped arrow means inhibit) so we would expect p16 to decrease in CDK4-inhibitor-resistant cells (if we want to continue replicating, we need CDK4, so we don’t want p16 to inhibit CDK4). On the other hand, if CDK4 is inhibited, we might rely on another CDK-cyclin pair to activate the traffic light. CDK2 and Cyclin E can do that, so I expect both of those downstream proteins to increase to accommodate for inhibited CDK4.
I expect to see the loss of p16 and Rb and an increase in Cyclin E, CDK2, and CDK6.
RESULTS
I am in the process of developing novel, palbociclib-resistant, DDLPS cell lines. These cell lines were grown over 6 months while being exposed to the drug. Resistant cell lines demonstrated decreased protein expression of Rb, p16, and CDK2. I also wanted to see what the initial response was to survive drug treatment. Interestingly, the response to palbociclib treatment was to increase CDK2, CDK6, Cyclin E, and decrease pRb—almost opposite from long-term acquired resistant cells. Nonetheless, clinically, patients with DDLPS that have lower p16 or Rb may be more likely to demonstrate resistance to palbociclib. Next-generation sequencing of DNA and RNA expression in parent and resistant lines are underway.
METHODS & DAY IN THE LAB ROUTINE
- MONDAY
- 7:00 AM – Cell Culture: Since cancerous cells rapidly divide, I have to remove some of the cells from their storage dish so that there’s room for them to continue to grow. This process of cell culture is called “splitting” because we split the population of cells. During this time, if I want to collect cells for experiments, I would do that here. Typically, if I don’t need to collect cells, the ones I remove are put in the waste. The ones I collect can be stored in the freezer. I take care of 4 cell lines and have three plates of each so I have 12 plates in total. I work in a fume hood so I have a sterile working place to prevent contamination of the cells.

- 8:30 AM – Prepare drug inhibition assay: My project assesses how cell lines react to a drug called palbociclib. We can place a select amount of cells in small wells of a plate and then treat each well with increasing doses of the drug to determine the concentration that kills half of the cells. This dose is called the IC50. We would expect resistant lines to have a higher IC50 than non-resistant cell lines. At this point, I would count how many cells I have in total so I can evenly distribute them in each well. I’ll let them grow and get used to their new environment for a day, then I’ll treat them with drugs.
- 9:30 AM – Quantify protein from cells: Now I can leave the fume hood and go back to my lab bench. I collected some cells at 7 AM so I’ll sonicate my cells which is like a hard-core blender to break open the cell membrane. I can then boil my smoothie of cells to unravel the protein structure. Then I’ll use a color-changing solution that turns dark purple in the presence of protein. The darker the color, the more protein there is. I can use known concentrations of protein to compare my samples to determine how much protein I have.

- 10:30 AM – It is important to know how much protein I have because if I want to compare the concentration of a certain protein (like CDK4), I need to make sure the total protein concentration of all my samples is the same. If they aren’t, then I can dilute some of the samples so that they are all the same. We will be preparing for an experiment that tells us how much of a specific protein we have in different samples. The first thing to do is prepare a gel. It feels very similar to Jell-o and is about 1.5 mm thick. The gel can be thought of like dryer sheets–like a matrix of fibers. Later on, I will place my proteins at the top of the gel and run electricity through the gel so the proteins move. The protein I look for can range between 16-250 kDaltons and this matrix will allow for smaller proteins to move faster through the matrix but will slow down the bigger proteins at the top so this gel will separate my proteins by size.


- 11:30 AM – Clean Up and Leave: Time to clean up my station, wash any dishes that I used, and prepare stock solutions for anything that may be running out.
- TUESDAY
- 7:00 AM – SDS-PAGE: The experiment that separates proteins by size is called SDS-PAGE. I’ll take the gel that I made on Monday and the samples that I want to look at and put the samples at the top of the gel. Then I’ll run electricity through it for about 2 hours. The samples have been stained with a dye and I also placed a dye marker at the top of the gel so I can track where the proteins are in the gel. The marker will show bands of different colors to act as a guide so I can see how far along the proteins are moving.

- 9:30 AM – Transfer the gel: I’ll then take the gel and transfer all the protein onto a membrane using electricity. This takes about 45 minutes.
- 9:45 AM – While I wait, I can treat the cells from Monday with drugs.

- 10:30 AM – Identify the protein I want to look at: I can identify a specific protein on my membrane by letting my membrane soak in a solution with a primary antibody against a protein like CDK4. The antibody will only interact with the CDK4 protein. I’ll let that solution sit overnight so we are sure that the antibody is attached to the protein.
- 10:45 AM – Clean Up and Leave
- WEDNESDAY
- 7:00 AM – I’ll take out the membrane and “wash” it by soaking it in another solution. I can then soak it in another solution with a secondary antibody that will attach to the antibody we placed on the membrane on Tuesday. This secondary antibody has a fluorescent marker on it.
- 9:00 AM – Scan the membrane in a dark room: Like photo film, we can soak the membrane in a fluorescence solution and then put it in a scanner so we can get an image of the protein concentration across different samples. This process is called Western Blotting.

- 10:00 AM – I have to repeat this process with a control to make sure I loaded the sample concentration of protein in all my samples. I’ll soak the membrane in a different primary antibody overnight.
- 10:30 AM – Clean Up and Leave
- THURSDAY
- 7:00 AM – Split the cells again. I’ll also treat the experimental plate with all the cells and drug concentrations with a color-changing solution that turns orange if cells are alive. I can interpret how many cells are alive at certain drug concentrations by how orange the solution turns.
- 9:00 AM – Repeat the washing and secondary antibody process and then scan
- 12:00 PM – Clean Up and Leave
- FRIDAY
- 12:00 PM – Attend lab meeting with my Principal Investigator (PI/mentor) and other students to share my results and listen to other students’ results.