The Anderson Lab is interested in how genetic diversity contributes to variation in species populations. To address these questions we utilize the major human fungal pathogen Candida albicans. C. albicans typically reside within the gastrointestinal tract of the host as a harmless commensal but overgrowth of the natural host niche can lead to debilitating mucosal infections and systemic bloodstream infections associated with significant mortality. C. albicans is capable of colonizing a wide variety of host niches that reflects significant phenotypic plasticity of the organism. Additionally, the C. albicans genome is highly dynamic, which allows significant genetic variation to exist across isolates and over evolutionary time. Two strains can differ by over 1% sequence divergence due to the accumulation of sequence variants. Additionally, C. albicans are highly tolerant of full ploidy shifts, the presence of supernumary chromosomes (aneuploidy), and long tracts of loss of homozygosity (LOH).
Genetic basis of phenotypic diversity
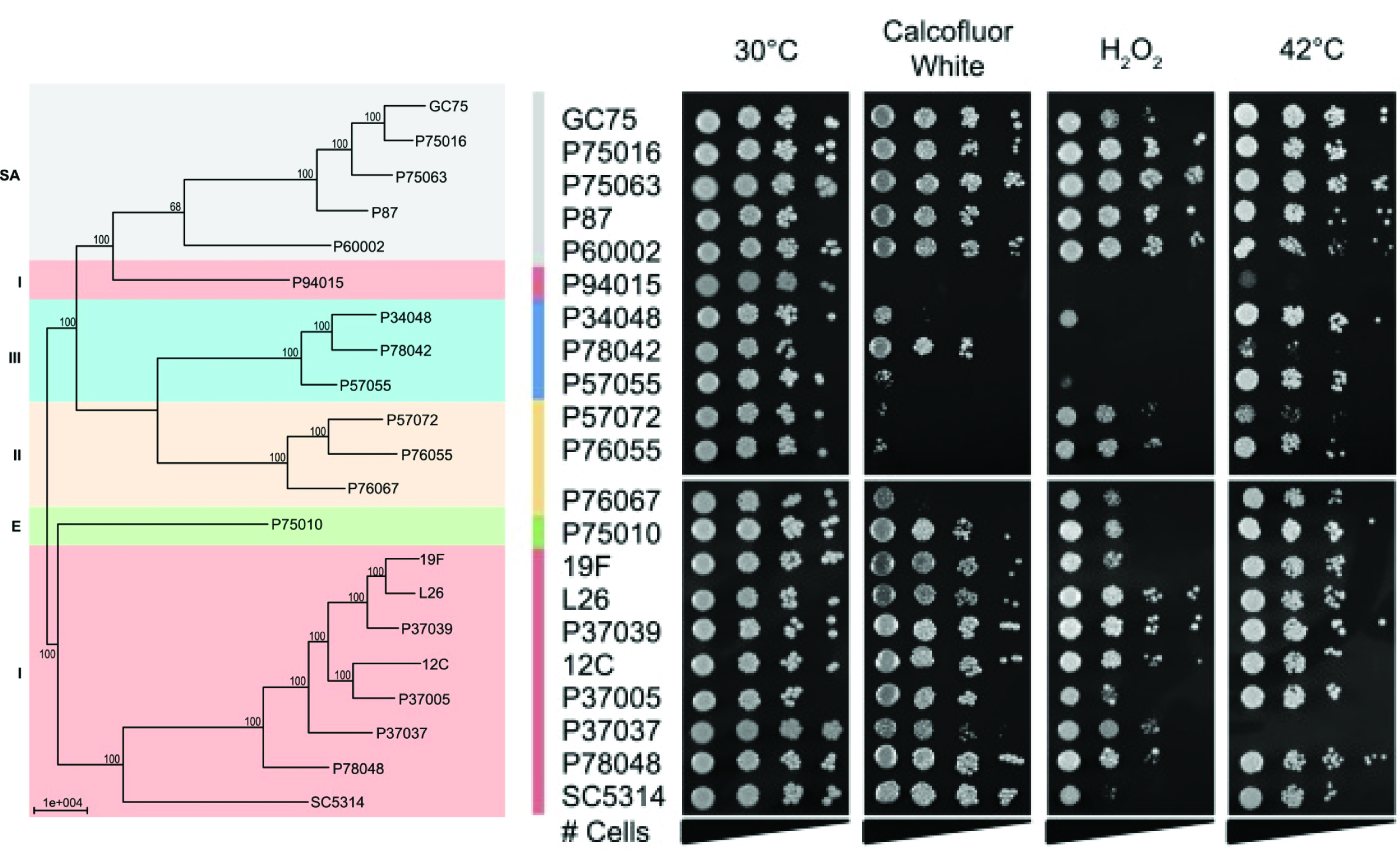
Widespread phenotypic diversity is found among C. albicans isolates for a range of phenotypes from growth rates and filamentation capacity to drug resistance and virulence in the host organism. Genotypic variation underlies much of this phenotypic diversity between isolates. We have developed tools and approaches to identify the contributions of genetic variants to this naturally-occurring phenotypic diversity in C. albicans. Existing polymorphisms form the basis for these studies and provide the opportunity to explore the genetic regulation underlying phenotypes of interest in an unbiased and comprehensive manner. Our lab combines informatics and experimental approaches to identify and test the genetic determinants of variation.
Expanded gene families in C. albicans

Gene duplication is the fodder for evolution to select for novel gene functions. The telomere-associated (TLO) gene family is the most expanded gene family in C. albicans compared to less pathogenic Candida species. These genes are paralogs of the Med2 tail component of the Mediator complex, a major transcriptional regulatory complex in eukaryotes. The TLO genes comprise three clades (α, β, and γ) based on sequence homology. However, it is unclear how individual paralogs contribute to specific phenotypes in C. albicans. We are investigating how gene diversification has shaped the molecular and biological role of individual genes during expansion.
TLO copy number and composition varies significantly between strains. Variation is likely due to rapid evolutionary dynamics that occur at chromosome ends and include nucleotide substitutions and large scale rearrangements. Passaging in vitro is sufficient to generate new Tlo proteins. The selective advantage of TLO variants and the evolutionary pressures acting on genetic changes is currently an active area of investigation.